Computational Chemistry
To effectively speed up your discovery process
Computational chemistry along with virtual screening plays an essential role in early Drug Discovery. Molecular modeling methods and screening technics become more and more sophisticated and together with machine learning are powerful tools for initial hit finding. The latest advances resulted in the drastic rise of Artificial Intelligence (AI) models, which now play a major role in every sector of pharmaceutical industry from early discovery to manufacturing and patient use.
Application of CADD (computer-aided drug design) in early stages allows significant savings on cost and time needed for data analysis after a large HTS campaign. Deep learning of protein structure and application of the latest calculation algorithms can enrich your hit rate up to 10-15%.
Being the largest supplier of Screening compounds and Fragments we extensively use all available computational chemistry tools and develop our own algorithms and approaches in hit finding. Using our experience we designed over 70 Targeted and Diversity libraries. Our experts participate in collaborations with leading scientific groups in computational chemistry field, confirmed by number of papers.
We will be happy to help you with in silico screening of our 3M Screening Collection or selected part from our REAL Database, as well as provide all the following CADD services:
- Virtual screening, Structure or Ligand-based. Docking calculation and 3D Pharmacophore search.
- Compound Library design considering all sensitive aspects of your project.
- Homology modeling of protein structures using AlpfaFold and search of potential binding sites.
- Molecular Dynamics (MD) simulations, protein structure and ligand interaction optimization.
- Activity analysis and QSAR modeling
- Design of hit follow-up libraries. Hit expansion and hit exploration.
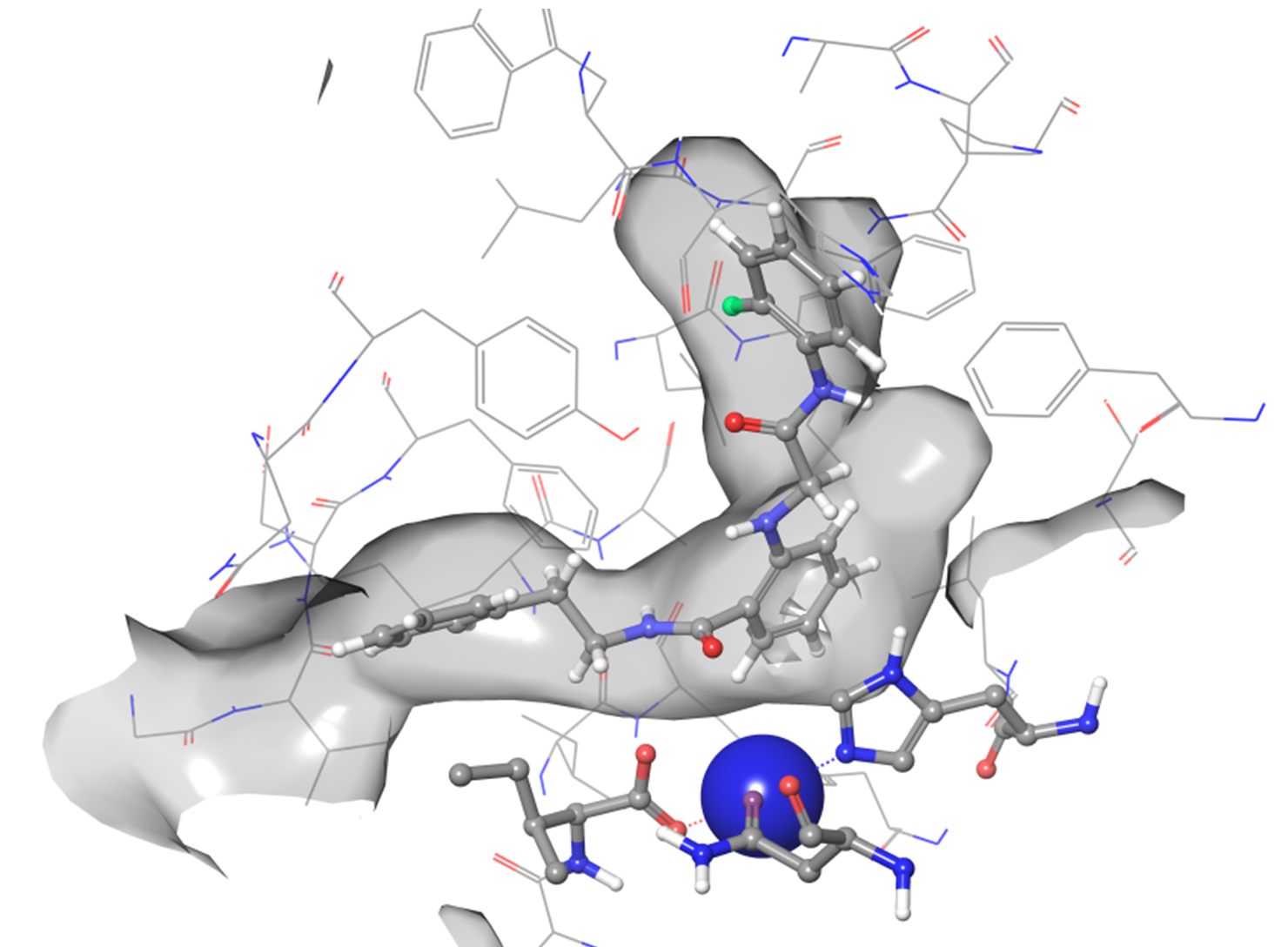
Lipoxygenase (hLOX-5) docking results: ligand binding pose of hit molecule.
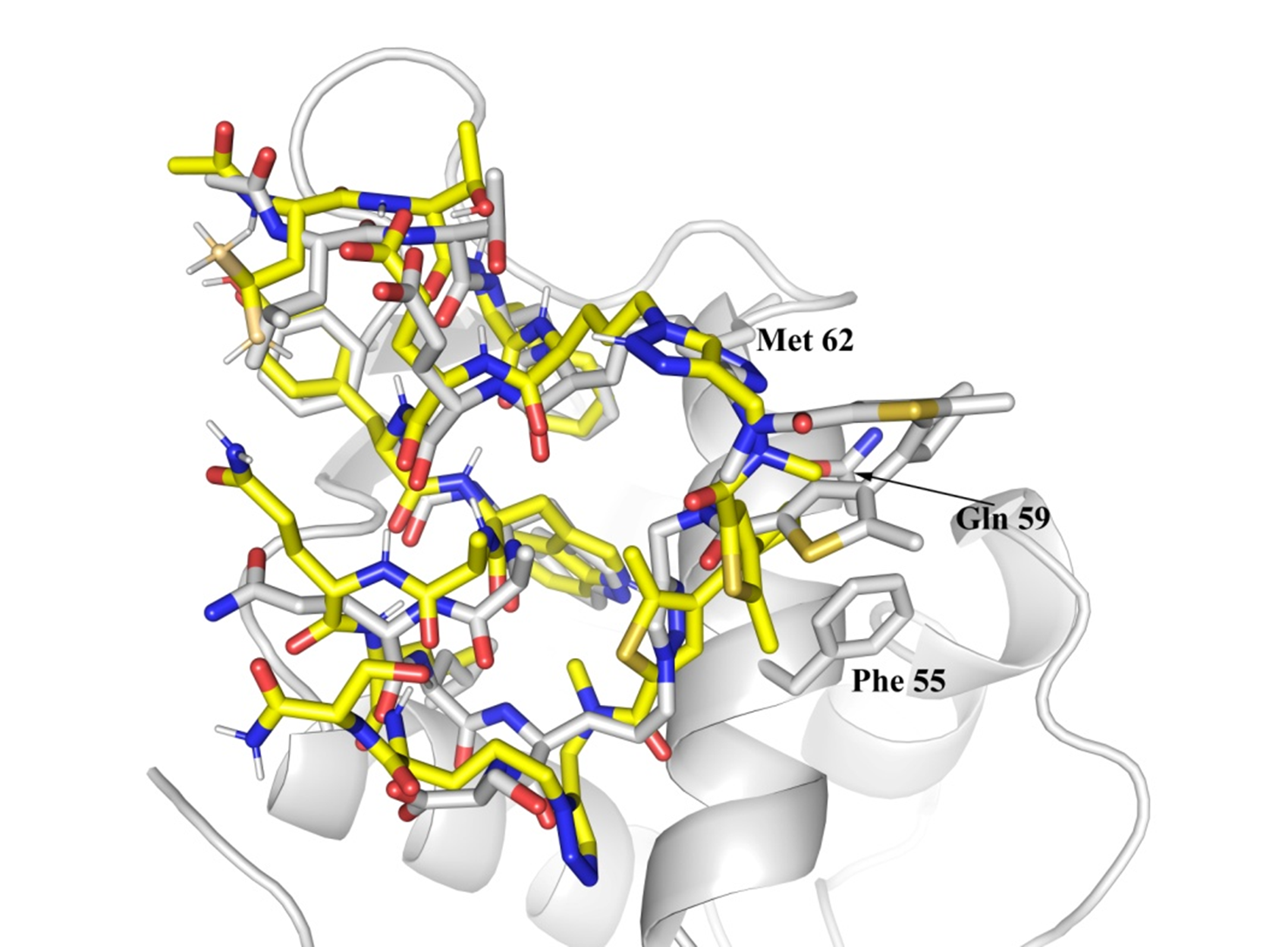
Structure optimization of stapled peptides and interaction with MDM2.
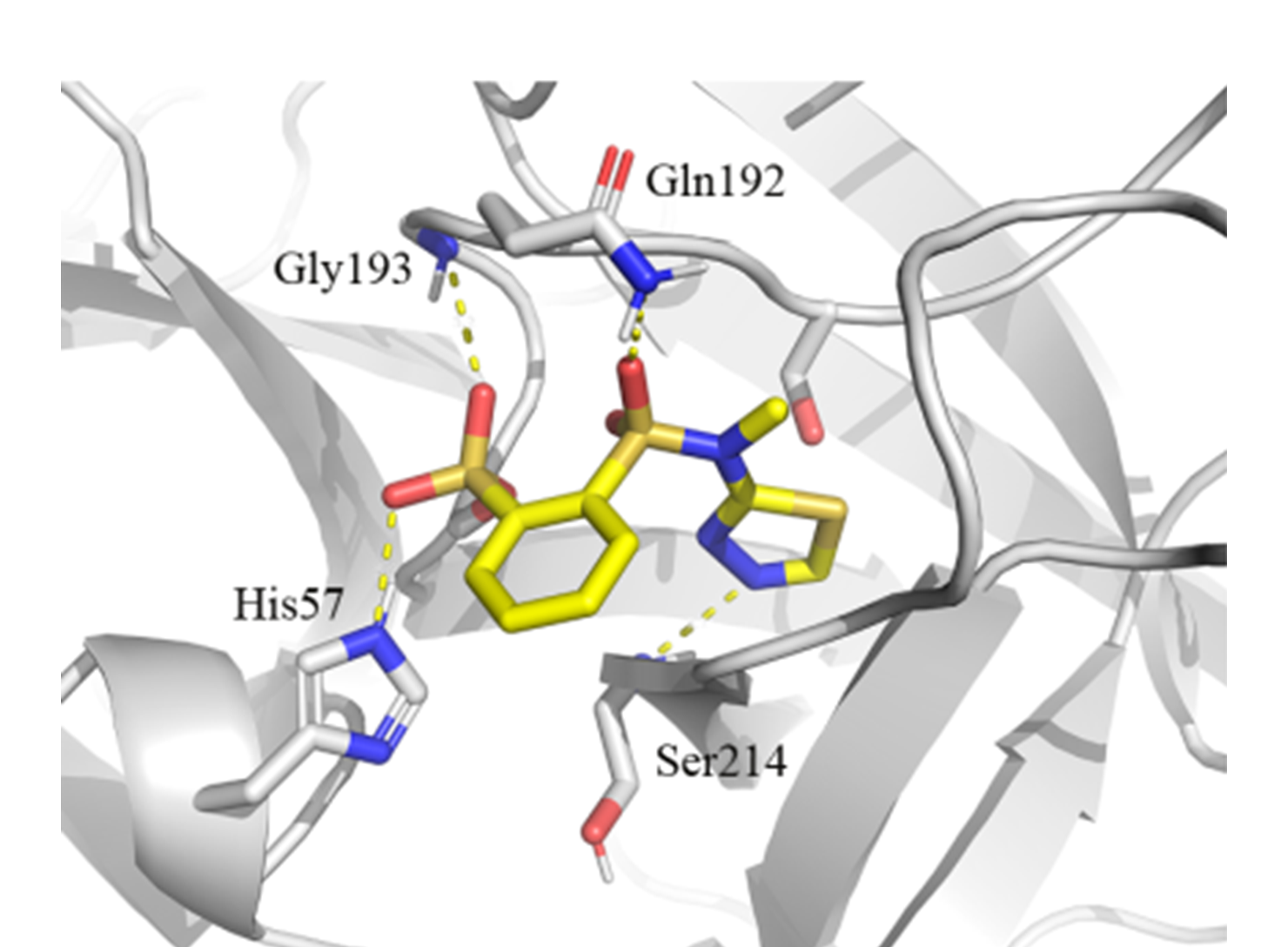
Covalent docking of Sulfonyl fluorides library against Serine protease.
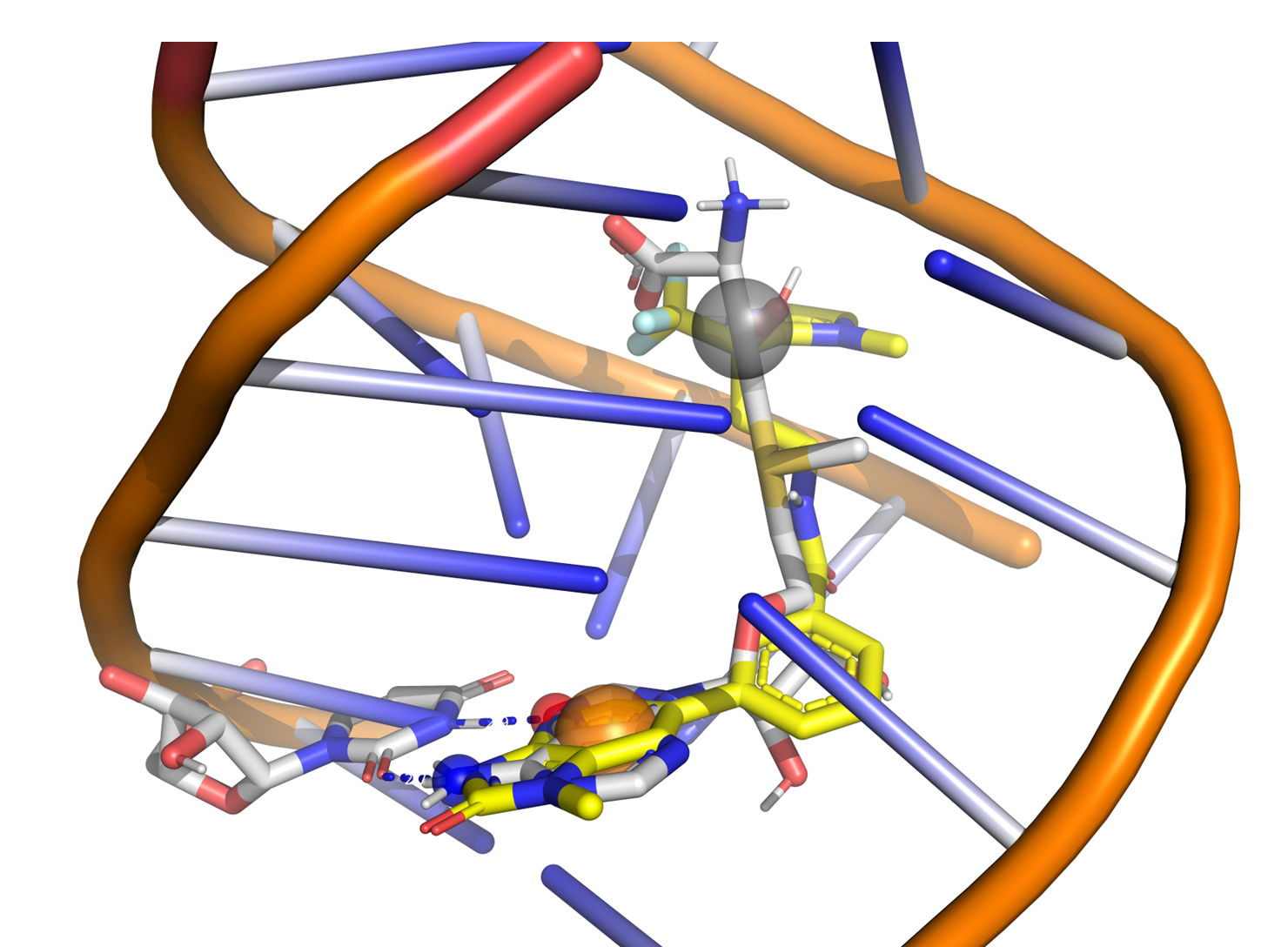
Molecular docking of RNA SAM riboswitch, example of identified hit binding mode (yellow).
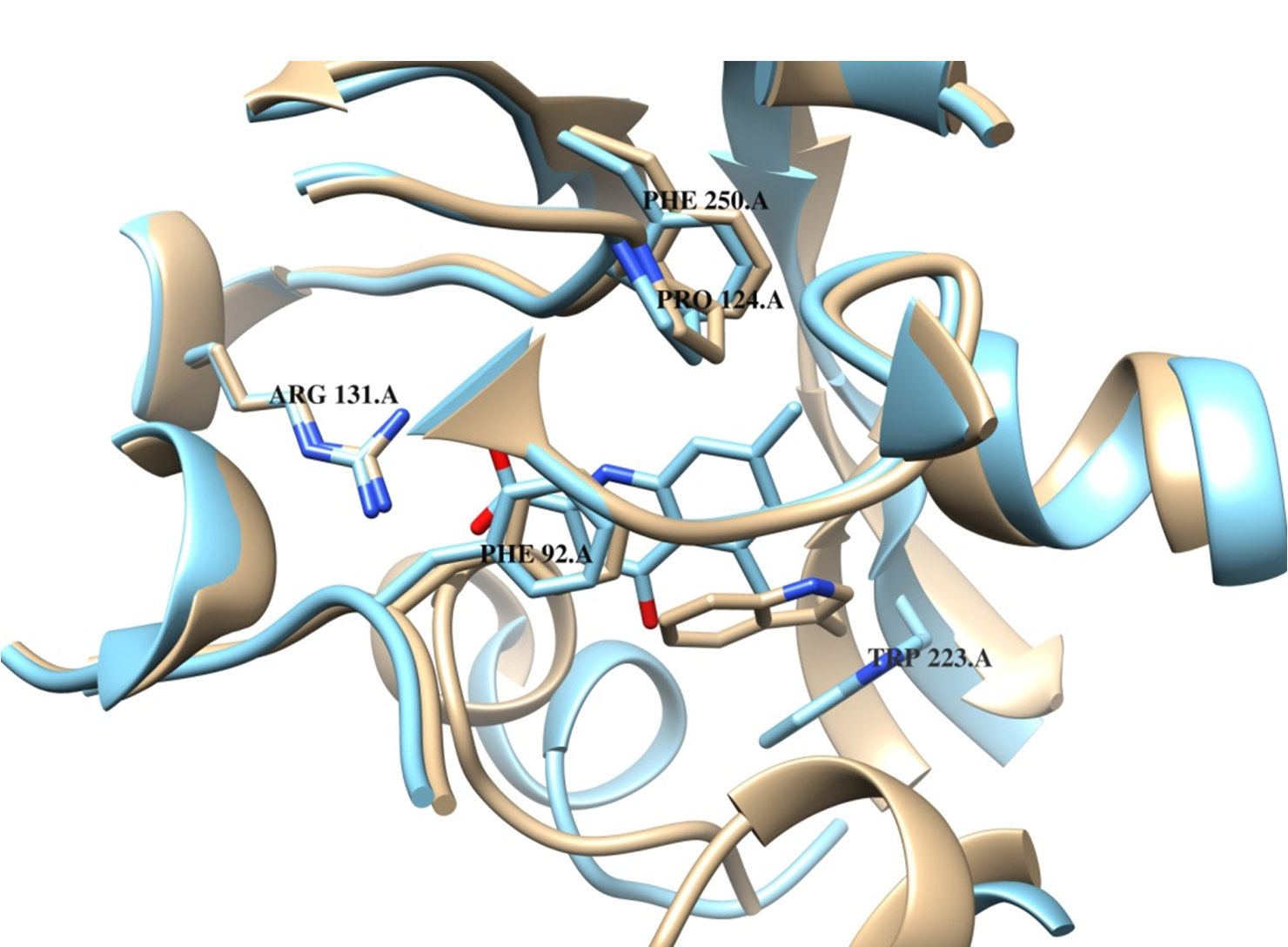
MD simulation and binding site identification for hNMDA based on the structure alignment with the reported liganded NMDA of rattuss norvegicus structure.
Identification of “hot-spots” as potential interaction sites for PPI inhibitors.
Selected publications
- Synthon-based ligand discovery in virtual libraries of over 11 billion compounds.
Nature 2022, 452-459. DOI: 10.1038/s41586-021-04220-9 - Generative and reinforcement learning approaches for the automated de novo design of bioactive compounds.
Chem. Commun. 2022, 129. DOI: 10.1038/s42004-022-00733-0 - Discovery of SARS-CoV-2 main protease inhibitors using a synthesis-directed de novo design model.
Chem. Commun. 2021, 5909-5912. DOI: 10.1039/D1CC00050K - An open-source drug discovery platform enables ultra-large virtual screens.
Nature 2020, 663-668. DOI: 10.1038/s41586-020-2117-z - Structure-based virtual screening and biological evaluation of novel inhibitors of mycobacterium Z-ring formation.
J. Cell. Biochem. 2022, 123, 852-862. DOI: 10.1002/jcb.30232 - Straightforward hit identification approach in fragment-based discovery of bromodomain-containing protein 4 (BRD4) inhibitors.
Bioorg. Med. Chem. 2018, 3399-3405. DOI: 10.1016/j.bmc.2018.05.010 - Diarylethene moiety as an enthalpy-entropy switch: photoisomerizable stapled peptides for modulating p53/MDM2 interaction.
Org. Biomol. Chem. 2020, 18, 5359-5369. DOI: 10.1039/D0OB00831A


